Why Teaching AI to Learn From Almost Nothing Could Change Drug Discovery
By Anoop Kumar Maurya | July 2026 | Series: AI for Drug Discovery — Article 1 of 10
 Image: Finding the right drug is like finding the one key that opens a lock — from a box of a million candidates
Image: Finding the right drug is like finding the one key that opens a lock — from a box of a million candidates
Imagine you are handed a completely new lock. No manual. No key. Just the lock — and a box of a million keys to try. Your job is to find the one that fits before the patient runs out of time.
That is drug discovery, roughly speaking. And for most of human history, we have done it almost exactly that way — trying key after key until something works.
This series of ten articles is about how artificial intelligence is changing that process. Not by magic. Not by replacing scientists. But by getting dramatically better at predicting which keys are worth trying before anyone bothers cutting them.
This first article covers the basics — what a drug is, what a protein is, what binding affinity means, and why the problem is harder than it looks. No equations. No code yet. Just the foundation you need before everything else makes sense.
💊 The One Sentence That Explains Drug Discovery
A drug is a small molecule that changes what a protein does inside your body.
That is it. Strip away everything else and every drug — aspirin, penicillin, insulin, a cancer immunotherapy — is doing exactly that. Finding a protein that is misbehaving and physically attaching to it to change its behaviour.
The rest of this series is about teaching computers to predict which molecules will attach to which proteins — and to do it well even when the data is scarce.
🧬 What Is a Protein, Actually?
Your body is run by proteins. They are the molecular machines doing almost everything:
- 🩸 Carrying oxygen through your blood (haemoglobin)
- 🍽️ Digesting your food (amylase, pepsin)
- ⚡ Firing your neurons (ion channels)
- 🔬 Replicating your DNA (polymerases)
- 🛡️ Defending against infection (antibodies)
A protein is built from a chain of amino acids — small chemical units, twenty different kinds in total, each with a single-letter code. A protein sequence looks like this:
MKTAYIAKQRQISFVKSHFSRQLEISFIASVQDNVQEPASTDRNMAKFTR...
That chain folds — automatically, in milliseconds — into a specific three-dimensional shape. And that shape determines everything: what the protein does, where drugs can attach to it, and whether a disease can exploit it.
🔄 The Sequence → Shape → Function Chain
Step 1: Amino acid sequence — the “recipe”
Step 2: Chain folds into a 3D structure — the “shape”
Step 3: Shape determines biological function — the “job”
Step 4: Disease disrupts that function → a drug restores it
This is why protein sequences matter so much in AI drug discovery. If you can read the sequence and predict the shape and function — without an expensive lab experiment — you have a massive head start.
💡 Fun Fact: The human body contains roughly 20,000 different proteins. Each one folds into a unique shape determined entirely by the order of its amino acids — a self-organising process that still surprises researchers fifty years after it was first described.
🔑 What Is Binding Affinity?
When a drug molecule encounters a protein, think of it like a key approaching a lock. Three things can happen:
| What Happens | What It Means | For Drug Discovery |
|---|---|---|
| Key does not fit at all | No binding | Useless compound |
| Key fits loosely | Weak binding | Needs optimisation |
| Key fits tightly | Strong binding | Promising drug candidate |
Binding affinity measures how tightly the drug and protein hold on to each other. The scientific unit is called Kd — the dissociation constant.
🔢 Understanding the Numbers
| Kd Value | Grip Strength | Real-World Meaning |
|---|---|---|
| < 1 nM | Iron grip | Extremely potent — best-in-class drugs |
| 1 – 100 nM | Firm handshake | Drug-like — most approved medicines |
| 100 nM – 10 µM | Loose grip | Weak — needs significant improvement |
| > 10 µM | Barely touching | Too weak to be useful |
💡 Why lower Kd = better drug: A lower number means the drug stays attached longer. A drug that falls off immediately cannot block or activate the protein long enough to have an effect.
The pKd Trick
There is one practical problem. Kd ranges from 0.001 nM to 10,000 µM — a billion-fold range. Neural networks struggle with numbers that vary that wildly.
So researchers convert it with a simple log transformation:
pKd = -log₁₀(Kd in molar units)
Now the range shrinks to roughly 5 to 12. Much easier to predict. When you see results reported as pKd values in the literature, this is why.
🔬 The Traditional Way: Try Everything
For decades, pharmaceutical companies solved the “which key fits” problem with brute force.
High-Throughput Screening (HTS)
 Image: Robotic arms running high-throughput screening — testing millions of compounds against a target protein
Image: Robotic arms running high-throughput screening — testing millions of compounds against a target protein
Step 1: Build a library of 1–10 million chemical compounds
Step 2: Use robots to test each compound against your target protein in tiny wells
Step 3: Measure which ones bind (fluorescence, radioactivity, or other assays)
Step 4: Keep the winners — usually just 0.01–0.1% of the library
This works. Many approved drugs were found this way. But the costs are staggering:
| HTS Reality | Numbers |
|---|---|
| Cost per screening campaign | $10–50 million |
| Time per target | 6–12 months |
| Hit rate (compounds that bind usefully) | 0.01–0.1% |
| Requires physical synthesis of compounds | Yes — before testing |
And here is the deeper problem: even when HTS works perfectly, it only works for proteins you can purify in sufficient quantities to run the assay. For rare targets with almost no prior research, that is another barrier entirely.
📊 What Machine Learning Changed
Researchers noticed something important. Every time a compound is tested against a protein in a lab, that result is data. And over fifty years of pharmaceutical research, millions of such results have accumulated in public databases:
| Database | Size | What It Contains |
|---|---|---|
| BindingDB | 2.4 million pairs | Binding affinities across diverse proteins |
| ChEMBL | 19 million activity records | Bioactivity data from published literature |
| Davis | 30,000 pairs | Kinase inhibitor selectivity |
| KIBA | 229,000 pairs | Kinase inhibitor bioactivity |
If you train a neural network on this data, it learns the pattern. Give it a drug molecule and a protein sequence — and it predicts their binding affinity. No lab required. No robots. Just a trained model and a few seconds of computation.
The best models — DeepDTA (2018) and GraphDTA (2021) — do this remarkably well. For certain proteins, their predictions are accurate enough to guide real drug discovery decisions at a fraction of the cost of wet-lab screening.
How Good Are These Models?
The standard accuracy metric is called the Concordance Index (CI):
CI = the fraction of drug pairs where the model ranks them correctly
- CI = 1.0 → Perfect ranking every time
- CI = 0.5 → Random guessing
- CI = 0.0 → Perfectly wrong every time
DeepDTA and GraphDTA achieve CI above 0.86 on standard benchmarks. That is genuinely impressive — and genuinely useful in a lab setting.
But there is a catch. A very large one.
💀 The Catch: The Data Desert
The databases these models train on are not balanced. They are heavily skewed toward a small set of popular, well-funded protein targets — mainly cancer-related kinases like EGFR and CDK2, which have been studied for decades and have thousands of known drug interactions.
Here is the uncomfortable reality:
Around 60% of human proteins have fewer than 10 measured drug interactions in any public database.
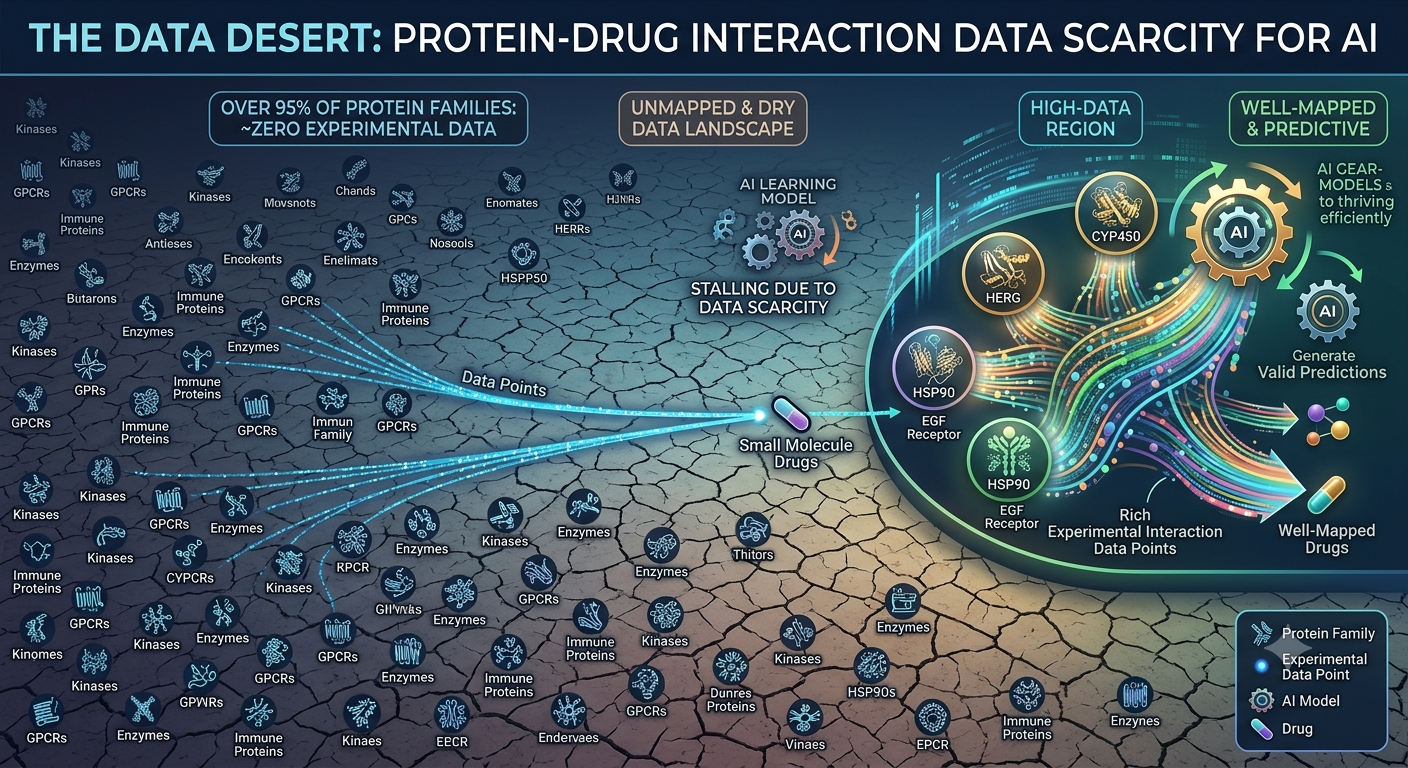 Image: The data desert — a vast majority of protein families have almost no drug interaction data for AI models to learn from
Image: The data desert — a vast majority of protein families have almost no drug interaction data for AI models to learn from
These neglected proteins are linked to:
- 🦠 Rare diseases — 7,000 rare diseases, most with no approved treatment
- 🔬 Newly discovered receptors — mapped by genomics but never studied in a drug context
- 🌍 Emerging pathogens — the next pandemic target, before any data exists
- 💉 Neglected tropical diseases — affecting hundreds of millions, underfunded for decades
What Happens in the Data Desert
When you point a state-of-the-art model at a data-scarce protein family, the results are stark:
| Scenario | Concordance Index | What This Means |
|---|---|---|
| Well-studied kinase family | 0.86 | Excellent — clinically useful |
| Under-represented protein family | 0.55 | Barely better than random |
| Random coin flip | 0.50 | Completely useless |
That gap — 0.86 versus 0.55 — is not a small technical problem. It means the most powerful computational tools in drug discovery are effectively unavailable for the diseases that need them most.
❌ Why Standard Deep Learning Cannot Fix This
The natural question: can we just train the model on the small amount of data we do have for rare targets?
No. And here is why.
A modern neural network has millions of parameters — numbers that need to be learned from examples. Learning them well requires seeing thousands of diverse examples. If you try to train on 10 examples, one of two things happens:
| What Happens | Technical Name | Why It Is a Problem |
|---|---|---|
| Model memorises the 10 examples perfectly, fails on everything else | Overfitting | Useless in the real world |
| Not enough signal to learn anything meaningful | Underfitting | Never gets off the ground |
Think of it this way:
Imagine trying to learn a language from ten sentences. You might memorise those ten sentences perfectly. But put you in a real conversation and you are completely lost — because you never learned the underlying grammar, only the surface patterns.
Standard deep learning needs thousands of examples. Rare protein targets have ten. This is the fundamental mismatch that drives everything in this series.
🧠 The Key Insight: Learn How to Learn
This is where the research in this series begins.
What if, instead of training a model to be good at one specific protein family, you trained it to be good at adapting quickly to any new family from very few examples?
Think about how an experienced doctor works:
A doctor who has spent years studying diverse diseases can diagnose a rare condition they have never seen before — by applying everything they know about symptoms, mechanisms, and patterns to the new case. Ten case studies is enough to get up to speed.
A computer program that memorised only one disease’s textbook cannot do this.
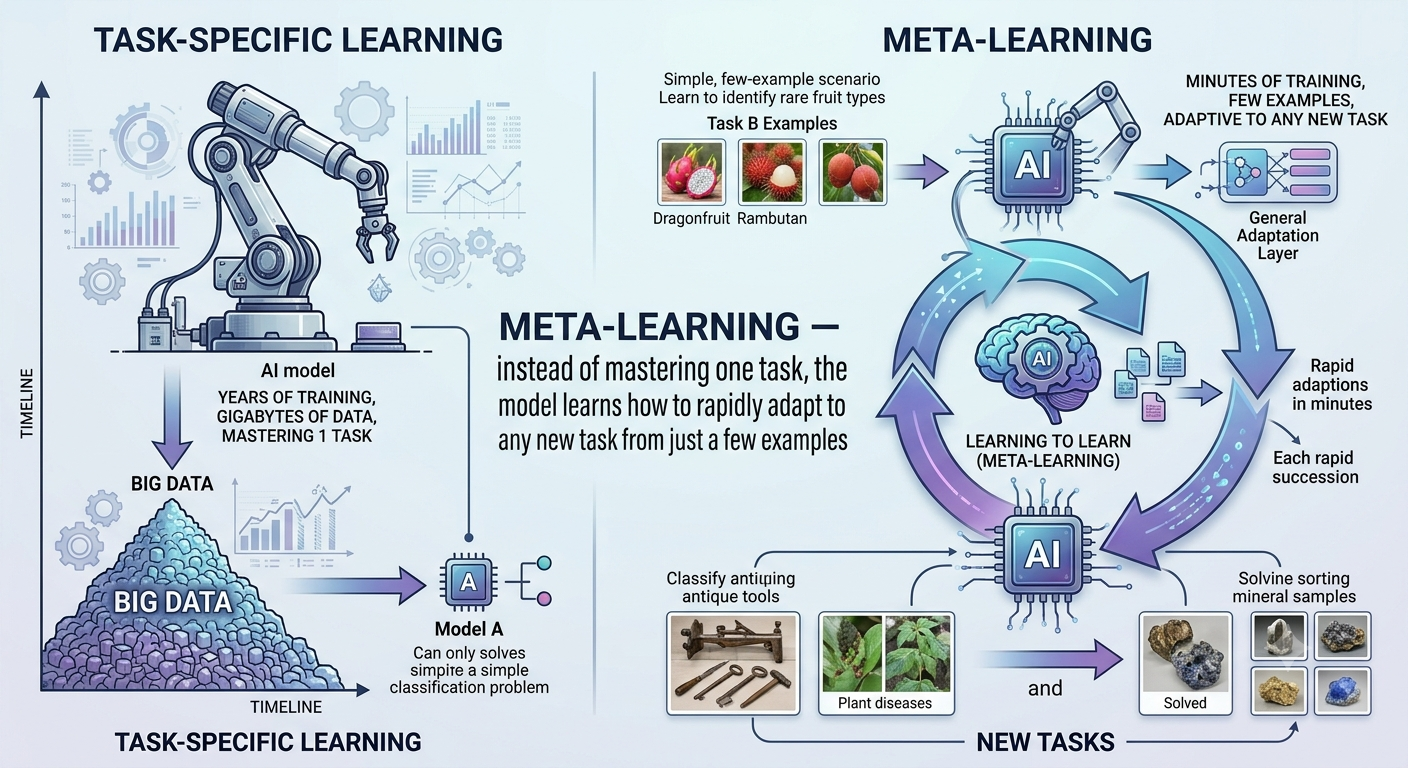 Image: Meta-learning — instead of mastering one task, the model learns how to rapidly adapt to any new task from just a few examples
Image: Meta-learning — instead of mastering one task, the model learns how to rapidly adapt to any new task from just a few examples
This idea — learning how to learn — is called meta-learning. Instead of training a model to be good at predicting binding affinity for kinases, you train it to be good at quickly becoming good at predicting binding affinity for any new protein family from minimal data.
🔄 Standard Training vs. Meta-Learning
| Standard Deep Learning | Meta-Learning | |
|---|---|---|
| Goal | Be good at one task | Be good at adapting to new tasks |
| Training data | Many examples from one task | Many examples across many diverse tasks |
| At test time | Apply fixed model | Fine-tune rapidly from few examples |
| Fails when | Data is scarce | Tasks are too dissimilar |
| Best for | Well-studied targets | New, data-poor protein families |
This is not a silver bullet. But for the specific problem of rare targets with minimal data, meta-learning is the most promising direction in the field — and it is what the FewShotDTA system, developed during my M.Tech thesis at IIT Patna, is built around.
📚 What This Series Covers
Over the next nine articles, we will build from these foundations to a complete, research-grade system — from biological intuition to working neural architecture to evaluation:
| Article | Topic | What You Will Learn |
|---|---|---|
| 01 (this one) | The core problem | Drug discovery AI, binding affinity, the data desert |
| 02 | Proteins deep dive | Sequences, structures, families, how AI reads them |
| 03 | Drug molecules | SMILES notation, molecular graphs, chemical features |
| 04 | Traditional DTA models | DeepDTA and GraphDTA — how they work and where they fail |
| 05 | Graph Neural Networks | How AI learns directly from molecular structure |
| 06 | Protein Language Models | How ESM-2 reads sequences the way GPT reads text |
| 07 | Few-Shot Learning | The core idea behind learning from minimal data |
| 08 | MAML | Model-Agnostic Meta-Learning — learning how to learn |
| 09 | FewShotDTA | The full system — architecture, training, evaluation |
| 10 | Results and next steps | What worked, what did not, and where this goes |
Each article builds on the last. No prior drug discovery knowledge is assumed — just a general comfort with the idea that neural networks learn from data.
💡 Who this series is for: ML practitioners and researchers who want to understand the drug discovery problem deeply enough to contribute to it. You do not need a biology background. You do need curiosity and patience with new domains.
🧩 Try This: Before You Read On
Take two minutes and try this thought experiment:
You are a new doctor. You have seen 500 patients with common diseases and can diagnose them accurately. Now a patient walks in with a disease you have never seen. You have 10 case studies from a medical journal.
How would you approach the diagnosis? What would you draw on? How is this different from looking something up in a textbook?
Write down your answer. Then, as you read Articles 07 and 08 on few-shot learning and MAML, come back to it. You will find that the meta-learning algorithm is doing almost exactly what you described — just with gradient descent instead of medical intuition.
✅ Before You Move On — Check Your Understanding
Try answering these without looking back:
- What is the difference between Kd and pKd? Why do we convert to pKd for machine learning?
- A model achieves CI = 0.55 on a new protein family. Should you trust its predictions for drug discovery decisions?
- Why can you not just train a deep learning model on 10 examples for a rare target?
- What does “learning how to learn” mean in plain English? How is it different from standard training?
If you can answer all four clearly — you are ready for Article 02.
📖 Key Terms — Quick Reference
| Term | Plain English |
|---|---|
| Drug | Small molecule that changes what a protein does |
| Protein | Chain of amino acids folded into a 3D molecular machine |
| Amino acid | One of 20 chemical building blocks that make up proteins |
| Binding affinity | How tightly a drug and protein hold on to each other |
| Kd | Dissociation constant — lower = tighter, longer-lasting binding |
| pKd | –log₁₀(Kd) — compressed scale from ~5 to 12, easier to predict |
| HTS | High-Throughput Screening — robotic brute-force drug testing |
| DTA | Drug-Target Affinity — the prediction task this series is about |
| Concordance Index (CI) | Fraction of drug pairs ranked correctly (0.5 = random, 1.0 = perfect) |
| Data desert | Protein families with fewer than 10 known drug interactions |
| Overfitting | Model memorises training examples and fails on new ones |
| Meta-learning | Training a model to adapt rapidly to new tasks from few examples |
Tags: drug discovery, AI, machine learning, proteins, binding affinity, deep learning, beginner, series